seqlm: an MDL based method for identifying differentially methylated regions in high density methylation array data
Abstract
Motivation: One of the main goals of large scale methylation studies is to detect differentially methylated loci. One way is to approach this problem sitewise, i.e. to find differentially methylated positions (DMPs). However, it has been shown that methylation is regulated in longer genomic regions. So it is more desirable to identify differentially methylated regions (DMRs) instead of DMPs. The new high coverage arrays, like Illuminas 450k platform, make it possible at a reasonable cost. Few tools exist for DMR identification from this type of data, but there is no standard approach.
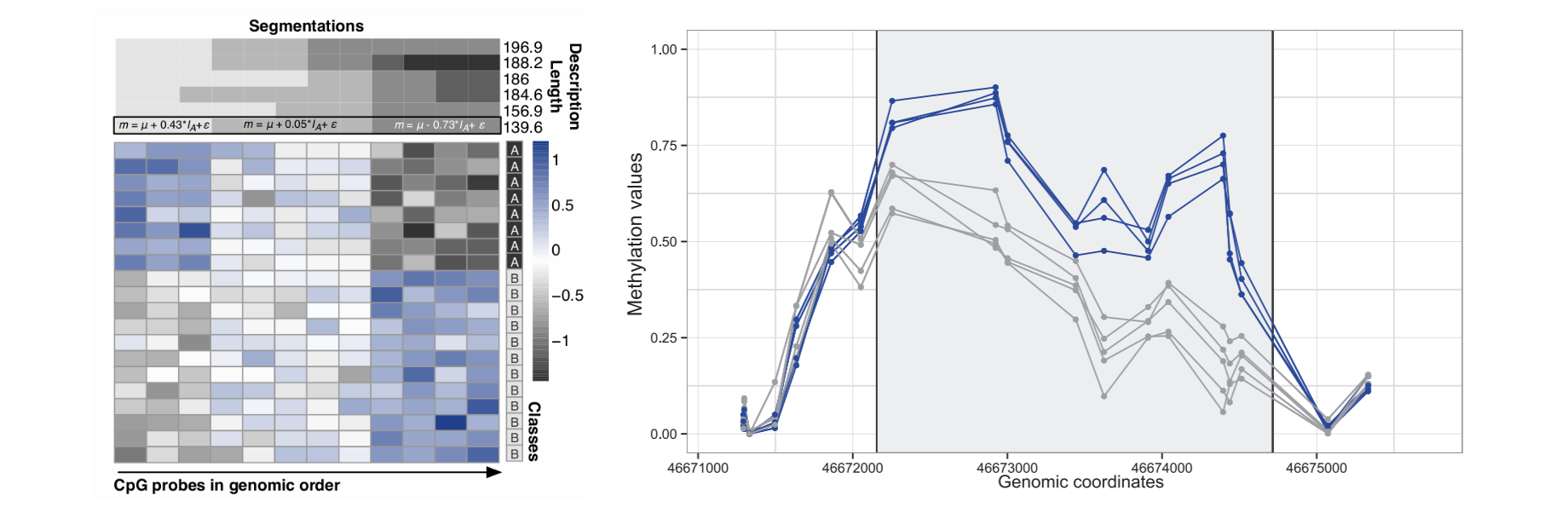
Results: We propose a novel method for DMR identification that detects the region boundaries according to the minimum description length (MDL) principle, essentially solving the problem of model selection. The significance of the regions is established using linear mixed models. Using both simulated and large publicly available methylation datasets, we compare seqlm performance to alternative approaches. We demonstrate that it is both more sensitive and specific than competing methods. This is achieved with minimal parameter tuning and, surprisingly, quickest running time of all the tried methods. Finally, we show that the regional differential methylation patterns identified on sparse array data are confirmed by higher resolution sequencing approaches.
Our R package is available on github